 Old Web
Old Web
HADHB

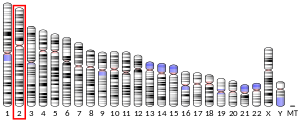
3032231086ENSG00000138029ENSMUSG00000059447P55084Q99JY0NM_000183NM_001281512NM_001281513NM_145558NM_001289798NM_001289799NP_000174NP_001268441NP_001268442NP_001276727NP_001276728NP_663533Trifunctional enzyme subunit beta, mitochondrial (TP-beta) also known as 3-ketoacyl-CoA thiolase, acetyl-CoA acyltransferase, or beta-ketothiolase is an enzyme that in humans is encoded by the HADHB gene. Trifunctional enzyme subunit beta, mitochondrial (TP-beta) also known as 3-ketoacyl-CoA thiolase, acetyl-CoA acyltransferase, or beta-ketothiolase is an enzyme that in humans is encoded by the HADHB gene. HADHB is a subunit of the mitochondrial trifunctional protein and has thiolase activity. The HADHB gene is located on chromosome 2, with its specific location being 2p23. The gene contains 17 exons. HADHB encodes a 51.2 kDa protein that is composed of 474 amino acids; 124 peptides have been observed through mass spectrometry data. This gene encodes the beta subunit of the mitochondrial trifunctional protein, a catalyst of mitochondrial beta-oxidation of long chain fatty acids. The HADHB protein catalyzes the final step of beta-oxidation, in which 3-ketoacyl CoA is cleaved by the thiol group of another molecule of Coenzyme A. The thiol is inserted between C-2 and C-3, which yields an acetyl CoA molecule and an acyl CoA molecule, which is two carbons shorter. The encoded protein can also bind RNA and decreases the stability of some mRNAs. The genes of the alpha and beta subunits of the mitochondrial trifunctional protein are located adjacent to each other in the human genome in a head-to-head orientation. Mutations in this gene, along with mutations in HADHA, result in trifunctional protein deficiency. Mutations in either gene have similar clinical presentations. Trifunctional protein deficiency is characterized by decreased activity of long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD), long-chain enoyl-CoA hydratase, and long-chain thiolase. This deficiency can be classified into 3 main clinical phenotypes: neonatal onset of a severe, lethal condition resulting in sudden infant death syndrome (SIDS), infantile onset of a hepatic Reye-like syndrome, and late-adolescent onset of primarily a skeletal myopathy. Additionally, some presents showed symptoms associated with myopathy, recurrent and episodic rhabdomyolysis, and sensorimotor axonal neuropathy. In some cases, symptoms of the deficiency can present as dilated cardiomyopathy, congestive heart failure, and respiratory failure. The deficiency has presented as hydrops fetalis and HELLP syndrome in fetuses. A compound heterozygous mutation of the HADHB gene can causes axonal Charcot-Marie-tooth disease, which is a neurological disorder, which shows that mutations in this gene can result in deficiencies that present in new forms not currently described. HADHB is a functional molecular target of ERα in the mitochondria, and the interaction may play an important role in the estrogen-mediated lipid metabolism in animals and humans. Additionally, HADHB has been shown to bind to the distal 3’ untranslated region of renin mRNA, thereby regulating renin protein expression. This article incorporates text from the United States National Library of Medicine, which is in the public domain.
- CCF Conference Analysis
- Map Galaxy
- Academic Report
- What's New