 Old Web
Old Web
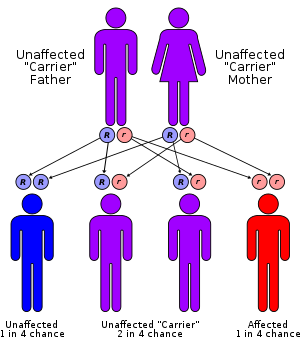
Usher syndrome

Usher syndrome, also known as Hallgren syndrome, Usher–Hallgren syndrome, retinitis pigmentosa–dysacusis syndrome or dystrophia retinae dysacusis syndrome, is a rare genetic disorder caused by a mutation in any one of at least 11 genes resulting in a combination of hearing loss and visual impairment. It is a major cause of deafblindness and is at present incurable.Ankyrin: Long QT syndrome 4 Usher syndrome, also known as Hallgren syndrome, Usher–Hallgren syndrome, retinitis pigmentosa–dysacusis syndrome or dystrophia retinae dysacusis syndrome, is a rare genetic disorder caused by a mutation in any one of at least 11 genes resulting in a combination of hearing loss and visual impairment. It is a major cause of deafblindness and is at present incurable. Usher syndrome is classed into three subtypes (I, II and III) according to the genes responsible and the onset of deafness. All three subtypes are caused by mutations in genes involved in the function of the inner ear and retina. These mutations are inherited in an autosomal recessive pattern. The occurrence of Usher syndrome varies across the world and across the different syndrome types, with rates as high as 1 in 12,500 in Germany to as low as 1 in 28,000 in Norway. Type I is most common in Ashkenazi Jewish and Acadian populations, while type III is rarely found outside Ashkenazi Jewish and Finnish populations. Usher syndrome is named after Scottish ophthalmologist Charles Usher, who examined the pathology and transmission of the syndrome in 1914. People with Usher I are born profoundly deaf and begin to lose their vision in the first decade of life. They also exhibit balance difficulties and learn to walk slowly as children, due to problems in their vestibular system. Usher syndrome type I can be caused by mutations in any one of several different genes: CDH23, MYO7A, PCDH15, USH1C and USH1G. These genes function in the development and maintenance of inner ear structures such as hair cells (stereocilia), which transmit sound and motion signals to the brain. Alterations in these genes can cause an inability to maintain balance (vestibular dysfunction) and hearing loss. The genes also play a role in the development and stability of the retina by influencing the structure and function of both the rod photoreceptor cells and supporting cells called the retinal pigmented epithelium. Mutations that affect the normal function of these genes can result in retinitis pigmentosa and resultant vision loss. Worldwide, the estimated prevalence of Usher syndrome type I is 3 to 6 per 100,000 people in the general population. Type I has been found to be more common in people of Ashkenazi Jewish ancestry (central and eastern European) and in the French-Acadian populations (Louisiana). People with Usher II are not born deaf and are generally hard-of-hearing rather than deaf, and their hearing does not degrade over time; moreover, they do not seem to have noticeable problems with balance. They also begin to lose their vision later (in the second decade of life) and may preserve some vision even into middle age. Usher syndrome type II may be caused by mutations in any of three different genes: USH2A, GPR98 and DFNB31. The protein encoded by the USH2A gene, usherin, is located in the supportive tissue in the inner ear and retina. Usherin is critical for the proper development and maintenance of these structures, which may help explain its role in hearing and vision loss. The location and function of the other two proteins are not yet known.
- CCF Conference Analysis
- Map Galaxy
- Academic Report
- What's New