 Old Web
Old Web
Neuroprotectin D1

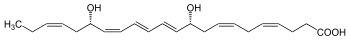
Protectin D1 also known as neuroprotectin D1 (when it acts in the nervous system) and abbreviated most commonly as PD1 or NPD1 is a member of the class of specialized proresolving mediators. Like other members of this class of polyunsaturated fatty acid metabolites, it possesses strong anti-inflammatory, anti-apoptotic and neuroprotective activity. PD1 is an aliphatic acyclic alkene 22 carbons in length with two hydroxyl groups at the 10 and 17 carbon positions and one carboxylic acid group at the one carbon position. Specifically, PD1 is an endogenous stereoselective lipid mediator classified as an autocoid protectin. Autacoids are enzymatically derived chemical mediators with distinct biological activities and molecular structures. Protectins are signaling molecules that are produced enzymatically from unsaturated fatty acids. Their molecular structure is characterized by the presence of a conjugated system of double bonds. PD1, like other protectins, is produced by the oxygenation of the ω-3 polyunsaturated fatty acid docosahexaenoic acid (DHA) and it is found in many tissues, such as the retina, the lungs and the nervous system. PD1 has a significant role as an anti-inflammatory, anti-apoptotic and neuroprotective molecule. Studies in Alzheimer's disease animal models, in stroke patients and in human retina pigment epithelial cells (RPE) have shown that PD1 can potentially reduce inflammation induced by oxidative stress and inhibit the pro-apoptotic signal, thereby preventing cellular degeneration. Finally, recent studies examining the pathogenicity of influenza viruses, including the avian flu (H5N1), have suggested that PD1 can potentially halt the proliferation of the virus, thus protecting respiratory cells from lethal viral infections. In vivo, PD1 is mainly produced as a response to inflammatory signals and it is found in various tissues, such as the retina pigment epithelial cells, lung epithelial cells, peripheral blood mononuclear cells (PBMC) and neural tissues. Studies in PBMC have shown that endogenous DHA, the main precursor of PD1, is released by the activity of phospholipase A2. According to these studies, PD1 is preferentially synthesized in PBMC cells skewed to the Type 2 T helper cell phenotype (TH2). This suggests that T-cell differentiation plays an important role in the activation of the PD1 biosynthetic pathway. The interaction of PBMC with interleukin 4 (IL-4), a potent inflammatory signal, leads to the differentiation of PBMC to TH2 type lymphocytes. In addition, activated TH2 cells further release IL-4, leading to the up-regulation of the enzyme 15-lipoxygenase-1 (15-LO-1). 15-LO-1 is a non-heme iron-carrying dioxygenase that adds oxygen atoms in a stereospecific manner on free and esterified ω-3 polyunsaturated fatty acids like DHA. Overall, the biosynthesis of PD1 proceeds through three distinct steps throughout which the activity of 15-LO-1 is essential. In the first step of the biosynthetic pathway, the binding of 15-LO-1 to its substrate (DHA) leads to the formation of the (17S)-hydro(peroxy)-DHA intermediate. This intermediate is rapidly processed to form a 16(17)-epoxide-containing molecule, which is the second intermediate. Finally, in the third step of the pathway, enzymatic hydrolysis of the 16(17)-epoxide-containing intermediate leads to the formation of PD1. In general, PD1 in vivo exhibits a potent anti-apoptotic and anti-inflammatory activity in the tissues in which it is localized. DHA, the main PD1 precursor, is mostly found in tissues such as the retinal synapses, photoreceptors, the lungs and the brain, suggesting that these tissues are more likely to be benefited from the protecting activity of PD1. RPE are essential in the survival and renewal of the photoreceptors in the retina. These cells exhibit a potent phagocytic activity that ensures the proper function of the retina. Therefore, oxidative stress can potentially damage the RPE cells and cause vision impairment. Studies in human RPE cells have suggested that the presence of oxidative stress triggering molecules, such as H2O2 causes the fragmentation of the DNA that in turn triggers apoptosis. These studies have proposed that PD1 acts as a signaling molecule and through its ligand-receptor interaction down-regulates the expression of genes, such as the transcription factor NF-κB. The inhibition of NF-κB results in the down-regulation of the pro-inflammatory gene COX-2 (cyclooxygenase-2) which is responsible for the release of prostaglandins, a potent pro-inflammatory mediator. In addition, PD1 has an important role in regulating the expression of the Bcl-2 family proteins (Bcl-2, Bcl-xL, Bax and Bad) that precedes the release of the cytochrome c complex from the mitochondria and the formation of the apoptosome. The presence of PD1 up-regulates the expression of the anti-apoptotic proteins Bcl-2 and Bcl-xL, while it inhibits the expression of the pro-apoptotic proteins Bax and Bad. Specifically, PD1 regulates this protein family by promoting the dephosphorylation of Bcl-xL by protein phosphatase 2A (PP2A) at residue Ser-62 which in turn heterodimerizes with the pro-apoptotic protein Bax and inactivates it. Consequently, the activity of the Bcl-2 family proteins results in the inhibition of the caspase 3 enzyme, thus preventing apoptosis and promoting RPE cell survival. Among others, Alzheimer's disease is characterized by the reduced concentration of PD1 and by the increased concentration of the amyloid-β peptide (Aβ42) that is responsible for the formation of senile plaques and also induces inflammation and apoptosis in neuronal tissues. Aβ42 is generated by the enzymatic cleavage of the β-amyloid precursor protein (βΑPP) through β- and γ- secretases. Like other pro-inflammatory mediators, Aβ42 induces inflammation through the activation of the pro-inflammatory enzyme COX-2 and the release of prostaglandins. Moreover, the release of Aβ42 down-regulates the anti-apoptotic proteins Bcl-2 and Bcl-xL and up-regulates the pro-apoptotic proteins Bax and Bad that ultimately lead to the formation of the apoptosome. PD1 in human neuronal glial cells (HNG) has been shown to trigger the down-regulation of βΑPP, thus decreasing the Aβ42 content in neuronal tissues and reducing inflammation and apoptosis. Specifically, PD1 in Alzheimer's disease models has been shown to respond to the increased concentration of the pro-inflammatory molecule Aβ42 by binding and activating the peroxisome proliferator-activated receptor gamma (PPARγ) either directly or via other mechanisms. According to some models the activation of PPARγ leads to increased ubiquitination and degradation of βAPP, thus reducing the release of Aβ42. Furthermore, PD1 inhibits the production of Aβ42 peptide by down-regulating β-secretase-1 (BACE1), while up-regulating the α-secretase ADAM10 and the secreted amyloid precursor protein-α (sAPPα). Overall, the above mechanism leads to the cleavage of βAPP protein though a non-amyloidogenic pathway that halts the formation of Aβ42 and prevents the premature neuronal degeneration.
- CCF Conference Analysis
- Map Galaxy
- Academic Report
- What's New